2
Data Extraction at the Margins of Health
The European Medicines Agency (EMA) approved forty-eight cancer drugs between 2009 and 2013. A study found that, at the point of approval, there was no evidence for improvement in either survival or quality of life in 65% of the different uses for which those drugs were approved. It seems that the EMA approved those drugs on the basis of hope, not evidence. As it turns out, hope was vindicated in only a minority of cases, because even after five years of follow-up studies, there was no evidence of improvement in either survival or quality of life in 53% of the different approved uses.1
There is nothing unusual about cancer drugs. For example, cholesterol-lowering statins are among the most widely prescribed of drugs, but meta-analyses have shown that they are only marginally effective at preventing heart attacks. Given the drugs’ relative weakness, in 2013 the American College of Cardiology and the American Heart Association released new guidelines aimed at increasing the drugs’ successes … by increasing the number of people taking them!2 In this context, it might be useful to remember that the ancient Greek word pharmakon can be translated as either ‘cure’ or ‘poison’. Or, in an adage attributed to the sixteenth-century alchemist and physician Paracelsus: ‘The dose makes the poison.’
Across almost all areas of medicine, close studies of recently approved drugs show that most drugs offer negligible new benefits. Prescrire, an independent healthcare evaluator, found that of ninety-two new drugs it evaluated in 2016, there were no breakthroughs, one ‘real advance’, five that ‘offer an advantage’, nine that are ‘possibly helpful’, fifty-six that offer ‘nothing new’, and sixteen that were ‘not acceptable’. Prescrire reserved judgment on five others. The 2016 results were not very unusual.3
The small benefits are connected to the high costs of trials. One doctor puts it this way: ‘Since [the pharmaceutical companies] anticipate that the drug will have little efficacy at best, affording slight benefit to most or more benefit to very few, the licensing trials are expensive, large, and sloppy.’ With so much at stake, these ‘[l]arge sloppy trials seeking small effects lend themselves to all sorts of data massaging and data torturing’.4
‘If you have to enrol a ton of people into your trial, that’s a sign the drug has a very small effect’, writes analyst Alan Cassels.5 Small expected effects push the companies to run ever-larger trials, enrolling ever more people. Estimates of the number of participants in clinical trials vary widely, but the number sits somewhere between three and six million annually.6
For pharmaceutical companies, extracting a statistically significant but small effect from a trial is much more important than shooting for a large effect. Most of the time, a small apparent effect in the data – usually in as few as two of the trials – is all that regulators demand for drug approval, and approval is the most important step in marketing the product. Most of the time, a small apparent effect is also all that pharma companies need to successfully sell their products.
‘A drug is a molecule surrounded by information’, I was told at a workshop for industry medical science liaisons that I attended in 2012. I would go slightly further: The right information surrounding the molecule makes it a drug. In particular, the right clinical trial information allows companies to make distinctions between their molecules and less effective ones, which brings regulators’ approvals and endorsements. Moreover, the right information allows companies to make strong claims about the drugs, which brings doctors’ buy-ins and recommendations.
As a result, running clinical trials has itself become an industry, one that serves pharma companies. I see it as a resource extraction industry: the trials extract fluids, measurements and observations from experimental bodies, to produce data. The data, after being heavily processed, becomes one of the key ingredients of a drug, crucial to bringing it to market and to making it circulate in that market. But the drug industry has evolved to be able to take advantage of very small effects, so most of the data extraction and processing happens at the very margins of health.
The Rise of Expensive Research
In the background of the pharmaceutical industry’s enormous influence over medical knowledge sit fifty-year-old changes in the importance of different kinds of medical research. Pressures from both government regulators and internal medical reformers have led to the rise of the randomized controlled trial (RCT) as the most valued and important kind of medical research. This change in the most important style of scientific reasoning7 in medicine has had huge effects. And the change is one that pharma companies have been well positioned to use to their advantage.
Medical Pressures
The RCT as a central plank of medical knowledge is relatively recent. In the English-speaking world, credit for the first real RCT in medicine is usually given to the UK epidemiologist Austin Bradford Hill, for his 1946 trial of the effect of streptomycin on tuberculosis, and for his advocacy of RCTs in medicine. One can find forerunners, such as Germany’s Paul Martini, who advocated for and performed RCTs on drugs starting in the 1930s, and gained influence in the 1940s.8 The RCT rose in importance over the following few decades to become the ‘gold standard’ of clinical research by the 1990s, in the wake of extensive advocacy by statisticians and medical reformers.9
For statisticians, random sampling in an experiment is the key requirement for making data amenable to statistical analysis. A well-designed and well-conducted RCT, by randomly assigning subjects from a population, produces results that have a defined probability of applying back to the population. Perhaps more important as a reason for the rise of RCTs, random sampling, especially combined with double blinding, has addressed widespread concerns in medicine about researcher bias.10 Since the 1950s, attempts to make RCTs the foundation of scientific medicine have been fairly successful, and since the 1970s physicians have been repeatedly told that RCTs are the only kind of reliable information on which to base clinical practice.
The rise of what is known as ‘evidence-based medicine’ has further promoted the idea that the practice of medicine should be based on RCTs – multiple RCTs, if possible. Evidence-based medicine started in the medical curriculum of McMaster University in Canada, based around practical clinical problem-solving. The clinical epidemiologist David Sackett led the way by developing courses on critical appraisal of the medical literature, which turned into a series of articles published in 1981.11 A decade later, on the invitation and patronage of Journal of the American Medical Association editor Drummond Rennie, those articles were updated and republished as a manifesto. The approach rejected doctors’ reliance on intuition – which had long been attacked – and even physiological reasoning and laboratory studies. The manifesto begins:
A new paradigm for medical practice is emerging. Evidence-based medicine de-emphasizes intuition, unsystematic clinical experience, and pathophysiologic rationale as sufficient grounds for clinical decision making and stresses the examination of evidence from clinical research.12
With this dense prose, the reformers were chronicling, applauding and promoting a revolution.
RCTs are not perfect tools, though. The artificialities of RCTs lead to knowledge that doesn’t map neatly onto the real world as we find it – the rigorously managed treatments in trials are rarely repeated in ordinary treatments, and populations studied are never exactly the same as the populations to be treated.13 In a related vein, RCTs tend to promote a standardization of treatment that does not fit well with the variability of the human world – in other words, the most effective standardized treatment may not be the most effective treatment for a particular patient in a particular context.14 In addition, most RCTs are worse at identifying uncommon adverse events than at showing drug effectiveness. Though RCTs are held up as a gold standard, poorly designed or executed RCTs may be of less value than sound versions of other kinds of studies.15 Illustrating all of the different ways in which RCTs are less rigid than they appear is the fact that studies supported by pharma produce much more positive results than do independent ones,16 showing that the method does not prevent bias.
Regulatory Pressures
Medical reform was one of the reasons why RCTs moved toward the heart of medicine. A second reason was the fact that government regulators made RCTs central to the approval process for drugs.
Much of modern drug regulation descends from the US Kefauver-Harris Act of 1962. Sponsors of the Act were responding to two sets of problems, though the Act did not actually address either of them. In the years leading up to the Act, Senator Estes Kefauver had put his energies into challenging the pharmaceutical industry on terrain where the US consumer had the most complaints: high prices stemming from patent-based monopolies. His efforts at reform were largely failures. Pharmaceutical companies and their industry association were able to deflect Kefauver’s attacks on drug patents and prices.17 The 1962 Act was spurred more directly by the compelling story of how the US had narrowly avoided disaster by not being quick to approve thalidomide – an episode used by the Kennedy Administration to push regulation forward.18 Dr Frances Kelsey of the Food and Drug Administration (FDA) had consistently questioned the safety of the drug, and had delayed its approval. Meanwhile, in Europe and elsewhere, thousands of babies had been disfigured by the use of thalidomide as an anti-nausea remedy and tranquillizer.
While the Act was supposed to improve the safety of drugs, in fact it added little to existing regulation of safety in the US. New was a requirement that drug companies show the efficacy of their drugs before they could be approved. Evidence of efficacy had to involve ‘adequate and well-controlled investigations’ performed by qualified experts, ‘on the basis of which it can fairly and responsibly be concluded that the drug will have its claimed effect’.19 The FDA structured its regulations around phased investigations, starting with laboratory studies and culminating in multiple similar clinical trials, which would ideally be RCTs.
It was only on the basis of the evidence from these trials that a drug could be approved for sale in the US and that claims for that drug could be made. The key provisions of the Kefauver-Harris Act concerned the appropriate and necessary scientific knowledge for the marketing of drugs. The FDA was already an obligatory point of passage for getting drugs to possible markets in the US.20 After the Act, though, approval also meant setting out the conditions for more marketing, by establishing what the companies could say about their drugs. The apparatus of approval, including laboratory and clinical research, became all about creating possibilities for advertising and otherwise promoting particular drugs.
The FDA had become a guarantor of sorts, offering a stamp of approval to assure doctors and patients that new drugs worked and weren’t too harmful. For the companies, this turned out to be a gold mine. Essentially, the FDA was vouching for the drugs it approved, and simultaneously limiting the competition. The approval system dramatically increased the value of patented drugs by adding layers of exclusivity.
Over the following few decades, regulators around the world followed the FDA’s lead, especially in using phased research culminating in substantial clinical trials as a model. For example, Canada’s regulations followed swiftly, in 1963. The United Kingdom established new measures that same year, and followed them up with a framework similar to the FDA’s in 1968. European Community Directives issued in 1965 required all members of the European Community to establish formal review processes, which they did over the next decade. Japan introduced its version of the regulations in 1967.
Especially since the expansion of drug regulation in the 1960s and 1970s, in-patent drugs have been promoted – and generally accepted – as more powerful than their older generic competitors. The result is that the drug patent has become a marker of quality, even though versions of most of the competitors were once patented, and often recently.21
In general, the drug industry has opposed new regulations, which increase costs, hurdles, and sometimes uncertainties.22 It also has challenged aspects of regulators’ authority in court.23 Drug companies and industry associations continually lobby regulators and legislators in quieter ways, to shape regulation in their interests around the globe.24 We can, for example, see industry interest in shaping regulation in the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). It is strongly in pharma companies’ interest to bring a new drug to market as quickly as possible, which increases the amount of time it can be sold while still under patent protection. Differences in the regulations for different major markets slow the process by requiring that the companies do tailored research to meet those different demands. For this reason, the International Federation of Pharmaceutical Manufacturers’ Associations created the ICH, bringing together the regulatory agencies of the European Union, Japan and the US.25 In a series of meetings beginning in 1991, the ICH standardized testing requirements, keeping the structure of phased investigations running from laboratory studies to RCTs, but ensuring that one set of investigations would be enough for these three major markets. There are many other ways, too, in which regulations are being standardized around the world. The many countries interested in pieces, however small, of the enormous pharmaceutical business bring their rules into line with those in place in North America and Western Europe, to make it easier for pharma companies.26
Though the industry complains about the costs of this research, and actively challenges the regulations that increase them, high costs have the unintended effect of preventing many non-industry researchers from contributing to the most valued kinds of medical knowledge: the RCTs of the kind that regulators require. Meanwhile, drug companies sponsor most drug trials, and in so doing affect their results. The companies fully control the majority of the research they fund, and to some extent they can choose what to disclose and how. Recent studies show that these companies don’t (despite being mandated to do so) publicly register all of the trials they perform, and don’t publish all of the data even from the trials that they do register.27 As I’ll show in the next few chapters, the articles they publish rarely make clear the full level of control the companies have had over the production of data, its analysis or its presentation; for example, company statisticians are rarely acknowledged, meaning that their contributions aren’t flagged.28 This allows the companies to use RCT data selectively to quietly shape medical knowledge to support their marketing efforts. At the same time, drug companies’ integration into medical research allows them to participate more overtly and broadly in the distribution of their preferred pieces of medical knowledge. The result is that drug companies have considerable control over what physicians know about diseases, drugs and other treatment options.29 So, while pharma companies have generally opposed new demands by regulators, they have also benefitted enormously from those demands.
Phased Research: An Outline
Bringing a drug to market involves many different kinds of research. Typically, it starts with a mixture of company strategizing, market research and biochemistry, where a company identifies a disease area in which it would like to have one or more products, and identifies some lines of biochemical or physiological research that look promising for that disease area.
Companies might identify initially promising substances – a process often given the somewhat misleading name ‘drug discovery’ – in any of a number of different ways, though the most common is by high-throughput screening. With a target human receptor in mind, the companies create an assay, a highly repeatable test for activity on the receptor. At that point, robots take over. Trays with between several hundred and several thousand copies of the assay are fed through a machine, which applies a different substance, from huge libraries of substances, to each copy of the assay. A good library might have nearly a million different substances, derived from soils, scraped from plants and moulds, or taken from anywhere else. The robot measures the effect of each of these substances on the assays, and promising candidates are marked for further rounds of testing, and eventually for pre-clinical studies.
With a smaller number of molecules in hand, the company begins learning about what they are, how they can be expected to act in human bodies, and how toxic they are likely to be. Laboratory studies follow, on both tissue samples and live animals. These lab studies are focused on learning how toxic and carcinogenic the molecules are, and so whether they are worth pursuing further. The choice of animal models – always including mice or rats, and one of dogs, pigs or primates – for different tests depends on the molecule’s expected form and effects, and is intended to gather information likely to be relevant to humans. Tests at this stage are structured by the ICH agreement, and results from these tests will be submitted to regulatory agencies as part of the application to pursue clinical tests in humans, and for eventual drug approval.
There are four different official phases of clinical trials for most drug applications (see Fig. 2.1). Phase I trials are tests on twenty to one hundred healthy human subjects. These trials, which I describe in more depth towards the end of this chapter, are designed primarily to determine the short-term safety of the candidate drug, but along the way the company will learn about its side effects, about comfortable dosage levels and tolerability, and perhaps something about efficacy.
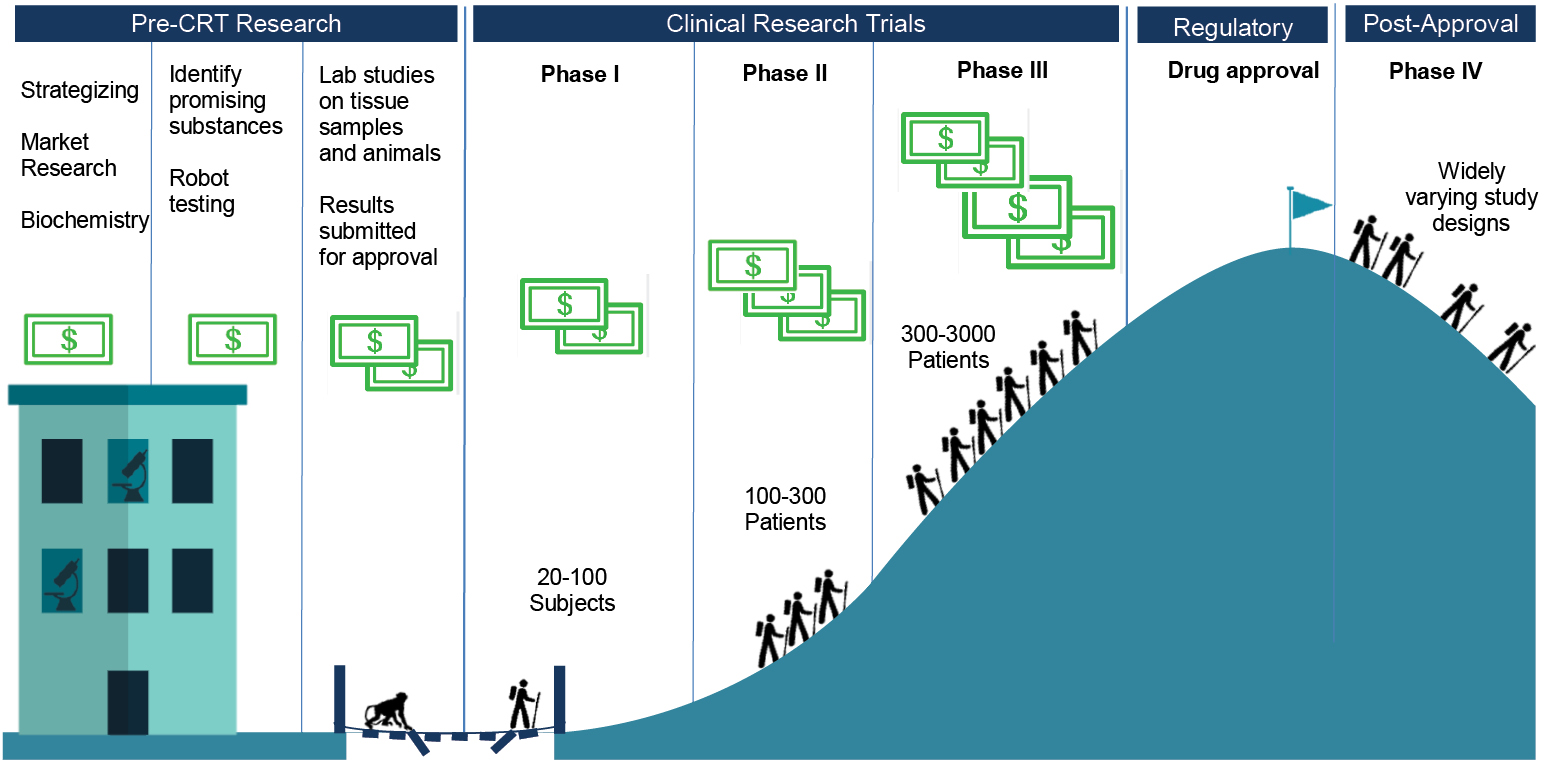
Fig. 2.1 Stages of drug development
Phase II trials bring in roughly one hundred to three hundred patients who have the targeted condition or disease. These trials are, therefore, to establish whether the candidate drug has biological effects that map onto medical treatment in a large enough percentage of the patients to make the drug worth pursuing – they should be RCTs, to allow for comparisons of treatment and placebo groups. Phase II trials also continue with the process of fine-tuning the dosage and monitoring safety.
At Phase III, the companies are running full trials designed to provide evidence of efficacy. They recruit populations that roughly match the ones for which they will be seeking market approval, and run trials that bear on how the candidate drug would officially be used, if approved. Depending on the condition to be treated, and the expected size of the drug’s effects, trials might recruit small or large numbers of patients, usually in the range of a few hundred to a few thousand. On the one hand, some conditions are too rare to recruit many patients, but on the other, if the drug is likely to show only marginal benefits, the number of patients in the trial has to be large enough to produce usable data. Phase III trials provide the key data feeding into applications to approve drugs, because if they are successful these trials provide evidence that the drug works, without causing too many negative effects. The evidence might not be in the form of cured patients, because many trials have only surrogate endpoints, markers taken to stand in for successful treatment – such as tumour size reduction in cancer, rather than extra years lived.
Finally, Phase IV trials include any clinical studies that take place after a drug has been approved. They might mimic trials of any of the earlier phases, depending on the purposes for which they have been designed. They might look like Phase I trials, if, for example, the goal is to provide evidence of whether a drug can be taken on an empty stomach. They might look like Phase III trials, if the goal is to provide fresh evidence to convince doctors to prescribe the drug. Or they might take yet different forms – for example, ‘seeding’ trials recruit doctors to prescribe a drug, to familiarize doctors with the product and to gather commercial information.
How Much Does Drug Development Actually Cost?
Industry-allied organizations – patient advocacy organizations, academic research units, think-tanks – can serve as a part of a larger echo chamber for the industry. One of the most important occupants of that chamber over the past forty years has been the Center for the Study of Drug Development (CSDD), located at Tufts University in the US. The CSDD was founded in 1976 by Dr Louis Lasagna, a prominent clinical pharmacologist known for his revision of the Hippocratic Oath. Lasagna was a key opinion leader who frequently worked closely with the pharmaceutical industry and had good connections with important US conservative figures.30 The CSDD is funded through grants from the industry, and gathers some its data directly from the industry, maintaining strict secrecy:
Data are collected from the people who create it – pharmaceutical and biotechnology companies. They cooperate because they know Tufts CSDD will generate a comprehensive and objective picture of the drug development process, while strictly ensuring that individual company data are not disclosed.31
The CSDD has had an enormous impact on the pharmaceutical industry, in particular through its regular estimates of the cost of developing a drug.32 By 2001, the estimate was a whopping $800 million, and by 2014 the CSDD had increased its estimate to an even more whopping $2.6 billion.33 Figures like these can be easily put to use to justify reduced regulation, longer monopolies, and especially high prices. In the face of $2.6 billion in costs, the industry contends, innovation could easily grind to a halt.
These figures are extremely controversial, to put it mildly. The data was provided by pharmaceutical companies on conditions of strict secrecy, so there is no way of knowing how representative it is. The studies included only drugs that the companies had ‘self-originated’, though probably the majority of drugs stem from publicly funded research. Research costs may include work that was unnecessary for approval. The figures take no account of tax incentives and other government subsidies. And the largest cost in the study – nearly half the total – is the opportunity cost of capital, calculated circularly, as if the companies invested in their own stocks (which have tended to increase in value enormously in recent decades, presumably in part because of drug development).34
Though competing estimates vary widely, and there is a fair amount of uncertainty about any estimate, a number of plausible figures come in at 10% to 25% of the CSDD ones.35 One recent study of publicly available data from US Securities and Exchange filings for firms developing cancer drugs, which duplicated the CSDD assumptions as closely as possible, came up with a figure of $648 million, still a hefty sum, but almost exactly 25% of the CSDD’s bloated figure.36
It may be that specific kinds of focused and efficient drug development projects can be much less expensive yet. The Drugs for Neglected Diseases Initiative, a non-profit originally founded by Doctors Without Borders, has developed seven new treatments for a mere $290 million.37 That works out at less than 2% of the industry’s claimed cost per drug. Because of this, and because the initiative is non-profit, it has been able to deliver treatments, including an antimalarial drug taken by 500 million people so far, at minimal costs. In 2013, when Andrew Witty, then CEO of GlaxoSmithKline, said that the CSDD figure was ‘one of the great myths of the industry’, he was suggesting that there were real efficiencies to be gained by focusing on projects more likely to be successful38 – the Drugs for Neglected Diseases Initiative seems to have taken that advice as far as possible. The projects it chose were unlikely to be profitable enough for any pharma companies to take on, so they were low-hanging fruit.
Integration of the Industry into Medical Research
Novelty, patents and regulation are bound up with an intensification of biomedical research. This has led to the accumulation and leveraging of what some scholars are calling ‘biocapital’,39 which involves the mutual cultivation of investment funds, biomedical infrastructure, and biological products and knowledge. We can see this even in public-private partnerships for drug development in the service of global health, with parties contributing in order to maintain claims on the circulating capital, materials and knowledge.40
Because of the expense of RCTs, companies and researchers have had to develop new formal structures to manage large trials.41 With large costs and overheads, the emphasis on RCTs has shifted the production of this most highly valued medical knowledge from independent medical researchers to pharmaceutical companies. The drug industry has steadily become more integrated into the medical research community, both because it produces important medical knowledge itself (generally through subcontractors) and also because it provides important funding for studies by more-or-less independent medical researchers.
Contract Research Organizations
Here is a standard image of pharma-sponsored research. First, a researcher designs and proposes a study. Then, looking for support, that researcher approaches one or more drug companies. A company may choose to fund the research, either out of interest in the results or to buy goodwill, or both. Then, the researcher performs the study, writes up a few articles on it and submits them to journals.
Journal articles stemming from industry-sponsored trials are likely to report conclusions favourable to the sponsors, as shown by multiple studies, systematic reviews and meta-analyses.42 For some areas within medicine, articles from industry-sponsored research are nearly certain to come to positive conclusions. In the standard image of sponsored research, then, we have to assume that more-or-less independent researchers align their studies with funders’ interests, through design, implementation, interpretation and/or publication. There are many ways they might do this. For example, the experimental drug can be tested against an inappropriate comparator or in an unrepresentative population; or, perhaps, the statistical tests are chosen with success in mind.
Though conflict of interest is a powerful force, it seems odd that conflicts of interest stemming merely from research funding could produce industry-friendly results in independent research, at least with the consistency that we see. Whether they are industry-friendly or not, we don’t have to believe that independent researchers are so easily influenced – because the standard image of sponsored research is wrong about what’s standard.
Pharma companies outsource almost all of their clinical research, some to academic groups but mostly to for-profit contract research organizations (CROs), which perform 70-75% of industry-sponsored research on drugs.43 The industry has been growing rapidly, at an average rate of 34% annually between 1997 and 2007, and at 15% annually in the years following. The growth is large enough that there are now trade associations for CROs; the UK-based Clinical & Contract Research Association, for example, boasts roughly thirty members, most of which are small or mid-sized CROs with primary offices in the UK. Since they produce research for hire, CRO science can represent drug company interests from the outset.
In 2014, I visited the European meeting of the Drug Information Association in Vienna. That conference is large, needing a convention centre to house the several thousand participants. At the Vienna meeting, speakers included drug company representatives, executives from CROs, regulators from a variety of different kinds of national and EU agencies, and patient advocates. It was an opportunity for these various stakeholders in bringing new drugs to the market to hear each other out – albeit in a formal setting, given the size of the meeting. Up for discussion was a broad array of issues about the design and regulation of clinical research, including issues about how to design regulations for testing and approving novel products, issues about how to plan clinical research so that it would feed into both approval and post-approval needs, and issues about transparency and disclosure. Although it was clear that there were divergences, almost everybody at the meeting had interests in the enormous project of the commercialization of drugs.
What struck me most was the exhibit hall, where CROs and other agencies working for the drug industry had set up slick booths to promote their services. There were many more CROs than I had known existed. Perhaps because the meeting was in Vienna, a number were emphasizing their abilities to run trials in Eastern Europe and the former Soviet Union, but their overall reach was global. Some CROs are specialized in other ways, like PaediaCRO, a German company that performs only paediatric research, or The Regulatory Affairs Company, an international firm that only handles drug approval submissions and the planning that goes into them. Many are full-service firms, ready to take on almost anything that drug companies don’t see a need to do themselves. Especially for small drug companies, outsourcing multiple functions makes sense, because they can then rely on CROs’ specialized knowledge and know-how.44
CROs can be involved at all stages of research: doing analytic and synthetic chemistry, performing in vitro and in vivo toxicology studies,45 providing laboratory services related to trials, running the trials themselves, analysing data, doing health economics research, pharmacovigilance (monitoring effects of drugs after approval), interfacing with regulators, and even handling the full drug approval application process.
Trials represent the core of the business, though. CRO-conducted trials are designed either for the drug approval process or for the further development of data to support the marketing of drugs, or for both. CROs, in turn, typically contract with clinics and physicians to do the hands-on work of clinical studies. They recruit patients in a variety of ways, through public advertisements, networks of specialists, or just through physicians’ practices.
Companies are becoming more sophisticated about populations, as well. In an interview in a study of offshored trials, a former Chief Executive of a CRO says, ‘Companies can now pick and choose populations … in order to get a most pronounced drug benefit signal as well as a “no-harm” signal.’46 This requires access to larger populations, often populations in multiple countries.
So, while CROs need to perform research of high enough scientific quality to support the approval and marketing of drugs, and they need to do so inexpensively and efficiently, they also need to serve other goals of the pharma companies that hire them.
CROs’ orientation to their clients leads them to make choices in the implementation and execution of the RCT protocol that are more likely to produce data favourable to those clients; they might, for example, skew the subject pool by systematically recruiting certain populations, or they might close some sites for breaches of protocol, especially if results from those sites are throwing up red flags. Given the enormous complexity of protocols for large RCTs, it would be no surprise if these choices contributed to the relationship between sponsorship and favourable outcomes.
Unlike the academics who are occasionally contracted to run clinical trials, CROs offer data to pharma companies with no strings attached. Data from CRO studies are wholly owned and controlled by the sponsoring companies, and CROs have no interest in publishing the results under their own names. The companies can therefore use the data to best advantage, as we will see in the next chapter. Company scientists and statisticians, publication planners and medical writers use the data to produce knowledge that supports the marketing of products.
Trials with and without Borders
CROs tend to work in multiple countries both within and outside North America and Western Europe, including poorer, ‘treatment naïve’ countries where costs per patient are considerably lower.47 However, North America and Western Europe still have more than 60% of the market share for drug industry trials. Of trials registered in the largest database, ClinicalTrials.gov, between 2006 and 2013 there were nearly ten times as many trials per capita in high-income countries as in middle-income ones.48 In terms of sites registered in this period – large trials are conducted at multiple discrete sites – nearly 70% of the nearly 460,000 were in North America and Western Europe.49 Nevertheless, an increasing number of sites are in Asia, Africa, South America and Eastern Europe; significant West-to-East and North-to-South shifts appear to be underway.50 The costs of trials, in terms of fees paid to physicians and subjects, and the cost of medical procedures, are much lower in the Global South, even in locations that have sufficient medical infrastructure to serve as good locations for trials.
India, for example, is well positioned to provide subjects. India’s Economic Times wrote in 2004, ‘The opportunities are huge, the multinationals are eager, and Indian companies are willing. We have the skills, we have the people.’51 India has invested heavily to establish the material, social and regulatory infrastructure needed to bring clinical trials to the country, for example, by providing education in the running of trials and establishing ethical standards.52 It is estimated that the costs per patient are from 30-50% lower in India than in North America or Western Europe.53
Government structures and incentives play a variety of roles. Seeing clinical trials as a major business, India has offered tax incentives to CROs and pharmaceutical companies for their local Research and Development (R&D), has dropped a requirement that clinical trials must have ‘special value’ to the country, has not insisted that experimental drugs later be marketed in India, and has invested in the training of clinical trial workers54 – key human resources in addition to trial subjects. As a result, all of the major international CROs have established operations in India, and there are also a large number of local CROs. For pharma companies looking for trial sites, there is a developing infrastructure in places like India.
Ethical variability is a further reason for the globalization of trials. Different standards apply even between established members of the European Union and newly admitted members in Eastern Europe. Even when participants in lower-income countries try to apply basic international ethical standards, circumstances still make for differences in practice. There have been cases of enormous differences in protocols between higher- and lower-income countries, which have sparked considerable debate; these extreme cases, though, are exactly that. CROs take more modest and careful advantage of ethical variability to lessen the cost and increase the efficiency of trials.55 CROs also compete with each other, creating downward pressure on the implementation of ethical standards; in this context, for example, industry actors complain about ‘floater sites’, pop-up research clinics whose short life expectancies create difficulties for CROs working with more established clinics.
To return to the case of India, Sonia Shah claims that the ethics infrastructure is weak in the country. She quotes health activist Sandhya Srinivasan as saying that ethics committees reviewing trial applications meet not to thoroughly review those applications, but ‘in order to enable clearance’.56 On the other hand, Kaushik Sunder Rajan argues that ethical frameworks and ethics committees are key to capacity-building for clinical trials in India; international companies operate on international standards, need trials to meet those standards and are ensuring the existence of ethics committees.57
There may also be public relations and liability pressures to move trials away from North America and Western Europe.58 Deaths and other severe adverse events are likely to attract more unwanted media attention in pharma companies’ core market areas than if they occur elsewhere. Legal liabilities tend to be much higher in North America and Western Europe, and especially in the United States, than in the rest of the world.
Why hasn’t pharma moved more quickly to lower-cost, lower-risk environments? Historical reasons are important. For example, before the ICH, the FDA insisted that the majority of trials used for a drug application be conducted in the US, resulting in the national development of material and social capital for running trials. In particular, Phase I trials are generally in-patient exercises, conducted in clinics with beds and other facilities; some of the older material infrastructure continues to be used. Just as there are in lower-income countries,59 there are even a number of established US and European populations of ‘professional guinea pigs’.60 Also, many countries are trying to attract trials: Denmark and others see their strong healthcare systems and ability to track individuals as offering good infrastructure for both recruitment and the running of trials.
But a significant part of the reason for the continued dominance of North America and Western Europe for Phase II, III, and IV trials is the importance of contacts with doctors in large markets. Clinical trials create and maintain those contacts. A large trial will often involve more than a hundred doctors, each of whom recruits a dozen or more patients. The doctors are paid both for recruiting patients and for all procedures that they, as investigators, perform as part of the trial. While they are earning that money, they are also becoming familiar with the drug and developing a stronger relationship with the company sponsoring the trial. Clinical trials can provide opportunities to sell drugs.
Investigators can also be enrolled to further help sell drugs once they are approved. As I describe in later chapters, they can become speakers for the company, giving talks to other doctors. If they are seen by the company as having the right kind of status, investigators can become authors on ghost-managed articles stemming from the trials. They can, in effect, become nominally independent advocates and salespeople for the drug being studied.
Investigator-Initiated Trials
If CROs take 70-75% of pharma’s trial business, what about the other 25-30%? What about the trials run by independent medical researchers but supported by drug companies? To find out, I set off to a large new Colonial Revival hotel in suburban New Jersey for a conference on these ‘investigator-initiated trials’ (IITs). Roughly a hundred pharma employees working on IITs attended, with a smaller number of people interested in selling services to the industry tagging along. This was a poor cousin of the Drug Information Association meeting I had attended in Vienna.
The companies support IITs both to further relationships with doctors and to contribute to positive scientific publicity. Sometimes, resonating with the term ‘IIT’, investigators will actually design trials and simply seek funding from companies with which they have established relationships. But more often, IITs are only partly independent. Mark Schmukler of Sagefrog Marketing, a general marketing firm with expertise in the health sector, writes that the goals of any IIT programme are:
- Adding to the base of knowledge for a product.
- Generating abstracts and publications to be shared with the medical community at congresses or meetings.
- Increasing the familiarity of key physicians with the use of a product.
- Producing advocates for the use of a product.61
Most importantly, ‘the IIT process itself, which derives from the Clinical Development Plan, should be timed carefully. For pre-launch trials, results and publications should come forward within 6 months of the anticipated launch’.62 All of this suggests that while IITs may be ‘investigator-initiated’ in some senses, they are still expected to fit neatly into the company’s marketing plans.
In fact, drug companies are inconsistent on these issues. At the meeting on IITs that I attended, panellists discussed technicalities of putting out requests for proposals for trials, starting from regulatory and marketing needs. Speakers and audience members explicitly recognized that if companies started from needs, especially regulatory needs, the trials couldn’t be fully independent – which ran afoul of some people’s ideas of what they were doing. One participant asked, ‘Doesn’t using a trial for drug approval mean a level of company involvement that means that it is a sponsored study?’
A senior medical affairs director for one company, Dr Moore, described how medical science liaisons need to ‘interact with investigators to get the right studies submitted to meet your corporate needs – without crossing lines!’ The trick is to make sure that investigators propose exactly what the companies need. ‘Say you need IITs in order to commercialize in a country …’, then you can work with influential doctors in the country, the relevant KOLs. Moreover, an advantage of IITs is that they tend to be much less expensive than CRO-run trials, especially when the trials take place ‘overseas’. Dr Macar, a vice president for medical affairs of a specialized drug company, referred to this as ‘outsourcing’. For Macar and Moore, these partly independent trials are a necessary part of commercialization.
Other drug company speakers, including one who had been working on IIT programmes for a number of years, were indignant about issuing requests for proposals and working closely with investigators to shape the trials. ‘We shouldn’t be doing any of these things’, insisted Mr Mayer, who went on to say that they can get the companies in trouble: ‘The last CIA [Corporate Integrity Agreement payment] for Pfizer was 3.2 billion [dollars]. It’s not funny anymore.’ Making sure that IIT programmes aren’t just a kind of ‘outsourcing’ helps to keep the companies safe from charges of manipulating evidence. Although the disagreement clearly wasn’t settled, Dr Macar tried to finesse a way through the uncomfortable problem. The distinction between control and independence in trials ‘still seems to be a pragmatic distinction, where it’s mostly grey’.
The Integration of Marketing and Research
John LaMattina, former President of Pfizer Global Research and Development, writes that ‘EVERY clinical trial carried out by the biopharmaceutical industry always has input from the company’s commercial division.’63 LaMattina doesn’t mean to say that the marketing department intervenes in a nefarious way, to turn good science into bad. Instead, he points to the fact that all clinical research is ultimately in the service of marketing drugs. Commercial goals shape the science:
Yes, biopharmaceutical companies have the goal of making profits. To achieve this, the billions of dollars that are invested in clinical trials must be judiciously spent. Companies draw on all experts in their organization (research, clinical, regulatory and commercial) to maximize the chances that the clinical trial, should it successfully meet its goals … will show the full value of the new medicine.64
The ‘full value of the new medicine’ is its maximum potential return for the companies. Research has to provide information that will define drugs and their markets, and will help move those drugs to their customers. To repeat a claim I made earlier in this chapter, a drug is a molecule surrounded by information. As a result, for pharmaceutical companies there is never a sense in which research is separate from marketing.
There are many ways in which drug companies can shape trials to make commercially useful outcomes more likely. Trials might involve advantageous comparator drugs, unusual doses, carefully constructed experimental populations, clever surrogate endpoints, trial lengths unlikely to show side effects, and definitions likely to show activity or unlikely to show side effects. In addition, companies can shape their publications of trials by including only some clinical endpoints, doing subgroup analyses, choosing advantageous statistical tests and presentations, heavily promoting positive results and burying negative ones, speculating about reasons for ignoring negative results or simply emphasizing positive results through the craft of writing. When Merck was testing its ill-fated painkillers, rofecoxib (Vioxx) and etoricoxib (Arcoxia) – these are COX-2 inhibitors, which should offer pain relief without the negative gastrointestinal effects of many traditional painkillers – it used every single one of these techniques to improve one or another of its published trials.65 We have more insights into these cases than most others, because of legal actions against Merck, but there is no reason to think that the company was doing anything out of the ordinary.
For example, when Merck ran a trial of its drug rofecoxib against an established painkiller, naproxen, it found more cardiac problems in the rofecoxib patients than in the control group. The company claimed that this was understandable, given that naproxen offered protection to the heart – even though naproxen’s heart benefits were entirely speculative. The initial journal article presenting that same trial’s results neglected to mention three of the heart attacks that occurred among the rofecoxib patients, because those heart attacks happened after the cut-off date for reporting them. Curiously, the cut-off date for cardiac adverse events was different from the cut-off date for gastrointestinal adverse events, which were expected to play out in favour of Merck’s drug, rather than the comparator drug. The resulting positive article was as heavily promoted as any piece of medical science could have been, because the company bought 900,000 reprints of the article to distribute to doctors.66
In the opening section of this chapter, I quoted a critical doctor accusing the industry of running ‘sloppy’ trials. The trials are only sloppy in the eyes of critics. For the companies, the trials can be very carefully designed to produce favourable results that can be used for market approval and then broader marketing efforts.
Everybody Is a Book of Blood67
Many of the participants in Phase I trials – trials on ‘healthy volunteers’ – have been through many such trials. Some even label themselves ‘professional guinea pigs’ or ‘professional lab rats’, because payments from trials make up most or all of their incomes. Typical studies pay between $2000 and $4000, though longer-term studies can pay significantly more. As a result, frequent participants hustle to be in the better studies.68
Most Phase I trials follow very similar protocols. The clinics recruit subjects by advertising, and the advertising is amplified through networks of experienced healthy volunteers. Subjects are screened to ensure that they are indeed healthy enough to participate, that they haven’t participated in another trial too recently, and that they aren’t taking any drugs that might interfere with the trial drugs. Frequent Phase I participants may lie in order to be eligible, and prepare their bodies so as to pass the screening tests – they can’t afford long washout periods between trials. The selected participants arrive on the first day of the trial and begin a daily regime of drug-taking, eating, sleeping and being subjected to a battery of tests: biopsies, drawing of blood, collection of urine and faeces, taking of vital signs, physical exams, and so on. Participants are restricted to the clinic, stay in dorm-like rooms, and move only from their beds to the cafeteria to common rooms to examining rooms. The food is standardized and measured out, and the days are routinized. Trials typically last from one to a few weeks, though there are shorter ones and the occasional much longer one. Sociologist Jill Fisher observes that risk is made ‘banal’ by the routines of Phase I trials, which are familiar to the frequent participants and clinic staff. Even the variations among trials follow familiar patterns.
Participants’ bodies are production sites. The participants ingest or are injected with novel substances, and then the clinic staff collect their tissues and fluids, and make less intrusive observations and measurements, to be turned into data. Blood is the key fluid: in addition to more routine draws of blood, many trials involve pharmacokinetic measurements that might involve ten, or as many as twenty, small draws on a single day, leaving the participants dizzy and nauseous.
Clinic staff monitor negative reactions to the drugs, including ones that pose real risks to participants. Fevers, nausea, vomiting, hallucinations and sleep paralysis would all be human events if they occurred outside the clinic; inside the clinic they are bodily reactions, and become data.69 Clinical trials are clinical in that they treat and observe actual bodies, but they are also clinical in their detached and unsentimental relationships with those bodies.
T.S. Eliot said, ‘The purpose of literature is to turn blood into ink’, and the same applies to clinical trials.70 Indeed, clinic staff refer to many Phase I trials as ‘feed ‘em and bleed ‘ems’ and there are routine references to ‘feeding and bleeding’, tasks that structure the days of staff and participants alike.71 Besides rhyming, the two tasks are related, because the feeding is a crucial step to allow bleeding. On the path toward the creation of medical facts, manipulated bodies are turned into marks on paper (and in computer files), allowing the unwieldy materials to be left behind and this newly created data to be neatly juxtaposed with other data.
Phase II-IV trials are quite different from Phase I trials, because they recruit subjects with the targeted health conditions or in the targeted markets, rather than ‘healthy volunteers’. They don’t pay participants more than honoraria, but they do offer some hope of treatment. They are more often outpatient trials than inpatient ones. They tend to be larger, sometimes much larger. And they follow much more varied protocols, so varied that it would be impossible to canvass them in a list.
But the vampiric experimental logic still applies to later-phase trials. Subjects’ bodies are still production sites. Subjects ingest or are injected with substances, and then their bodies are monitored for reactions. Blood is drawn, measurements are taken, outcomes are recorded. Negative outcomes and adverse events become data, treated clinically by the trial apparatus.
Conclusions
In the second half of the twentieth century, medical reformers successfully demoted the art of medicine in favour of the science of medicine. Meanwhile, policy reformers successfully focused drug regulators’ attention on scientific evidence of efficacy and safety. In both settings, the science in question has become dominated by clinical trials, preferably randomized controlled trials.
To work with these new regimes, pharma has become increasingly dependent on science. Large RCTs provide the data that regulators demand, data to show that a would-be drug has at least some more efficacy than a placebo, and that it isn’t too dangerous. Parallel and harmonized regulations around the world define bars for drugs to clear, and the industry invests exactly the resources needed to clear those bars. In return, national and international regulators – the most important elements of the market – vouch for the drugs and maintain a low-competition environment.
The pharmaceutical industry cannot, in general, rely on academics and academic institutions to run its growing numbers of clinical trials. Too much rides on timely success. For that reason, the industry turns to companies that often sit unnoticed in its shadows: contract research organizations. The CROs running most of the industry’s trials draw fluids, measurements and observations from the bodies of millions of participants to create one of the industry’s most precious substances, namely the data that, when processed, can turn an experimental substance into a marketable drug.
But much of this resource extraction sits at the margins of health. Most new drug applications show that only a few of the trials conducted find a statistically significant difference between the drug and a placebo. Most applications aren’t able to show a consistent medically meaningful effect. However, most of the time that small apparent effect in the data is all that’s needed for approval and to make positive cases to doctors.